
Peutz-Jeghersi sündroom on geneetiline haigus, mille esimene sümptom on sageli seletamatud nahamuutused. Millised selle haiguse sümptomid on veel? Miks on varajane diagnoosimine nii oluline? Millised on Peutz-Jeghersi sündroomi juhtimisstrateegiad?
Peutz-Jeghersi sündroom on geneetiline haigus, mis on pärilik domineerivalt autosomaalselt, mis tähendab, et haigusega inimesel on haige lapse saamise võimalus vähemalt 50 protsenti.
Peutz-Jeghersi sündroomi kirjeldused ilmusid kirjanduses aastast 1896, kuid selle põhjus leiti alles 1998. aastal.
Enamikul patsientidest on 19. kromosoomis LKB1 supresorigeeni mutatsioon, selle geeni produkt osaleb rakkude jagunemises ja vastutab rakkude polarisatsiooni eest. Meeskonda kuuluvad:
$config[ads_text1] not found- läätseplekid
- polüübid, eriti peensooles
- vähirisk on samuti suurem.
Peutz-Jeghersi sündroom: sümptomid
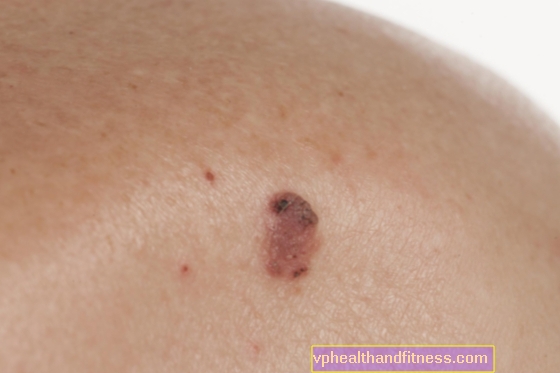
Läätseplekid (lad. lentigo) ilmuvad nahale ja on pigmenteerunud kahjustused, on heledad või tumepruunid, harvemini tumesinised, hästi piiritletud, mõne millimeetri suurused ning ümmarguse või ovaalse kujuga.
Läätseplekid tekivad tavaliselt lapsepõlves ja võivad noorukieas kaduda.
Läätseplekid on põhjustatud melanotsüütide (värvaine sisaldavate rakkude) paljunemisest epidermis. Peutz-Jeghersi sündroomi korral esineb neid arvukalt ja need asuvad tavaliselt suu, nina, silmade ümber, mõnikord ka põskedel, suus või kätes ja jalgades.
Kuigi läätseplekid on täiesti kahjutud, on need haiguse kõige nähtavam sümptom.
Need on patsientide seas kõige levinum ärevuse põhjus ja arstiabi otsimine ning need on diagnoosi allikad, mis viib õige diagnoosini.
$config[ads_text2] not foundPeutz-Jeghersi sündroomi teine eripära on polüübid.Üldiselt on need struktuurid seedetrakti limaskesta väljaulatuvad osad, see vorm võib omandada muu hulgas neoplasme, aga ka näiteks põletikulisi protsesse.
Peutz-Jeghersi sündroomi käigus on meil tegemist nn hamartomaatiliste polüüpidega. See üsna keeruline nimi tähendab vähkkasvajat, mis on arenguhäire.
On iseloomulik, et need polüübid koosnevad küpsetest kudedest, mida tavaliselt leidub kehas, kuid need ilmuvad kas vales kohas või on kaootiliselt jaotunud, näiteks soolestikus esineva polüpi korral ilmub kopsukude.
Hamutomaatilised polüübid Peutz-Jeghersi sündroomi käigus esinevad kõige sagedamini peensooles, peamiselt tühisooles, kuid need võivad ilmneda seedetrakti mis tahes osas: käärsooles, maos ja pärasooles.
Nende struktuuri iseloomulik tunnus on pikk vars. Üldiselt on polüüpide olemasolu asümptomaatiline ja kahjutu. Mõnikord on neil siiski obstruktsiooni oht, kuna need võivad seedetrakti valendiku täielikult blokeerida, põhjustades nn intussusception.
Samuti juhtub, et nad amputeerivad ennast või "purunevad", mis omakorda võib põhjustada seedetrakti verejooksu ja aneemiat.
$config[ads_text3] not foundPealegi on Peutz-Jeghersi sündroomi korral esinevad soolenähud kõhukinnisus ja kõhuvalu.
Kasvajad Peutz-Jeghersi sündroomi korral
Selle haiguse peamine probleem on asjaolu, et paljud polüübid võivad muunduda ja põhjustada vähki. Hinnanguliselt areneb umbes 50% (mõnede allikate hinnangul isegi 90%) selle diagnoosiga patsientidest vähk, enamasti soolestik, kuigi sagedamini esinevad ka rinnakasvajad, kopsuvähk, maovähk, kõhunäärmevähk, munasarjavähk ja emakavähk.
Mõned kasvajad võivad olla hormonaalselt aktiivsed ja põhjustada menstruaaltsükli häireid, meeste günekomastiat ja enneaegset puberteeti.
Peutz-Jeghersi sündroom: diagnoos
Diagnoosimine on võimalik, kui kõik kolm tingimust on täidetud:
- haiguse esinemine perekonnas ja selle pärimise viis - see määratakse meditsiinilise intervjuu põhjal
- naha ja limaskestade liigne pigmentatsioon
- polüüpide esinemine peensooles histopatoloogilise kinnitusega, et need on hamartomaatilised polüübid
Kui kallim on haige, on diagnoosimine palju lihtsam ja polüüpe pole vaja hinnata. Kuid alati on vaja välja jätta muud haigused, mis võivad põhjustada sarnaseid sümptomeid, eriti muud geneetilised sündroomid.
Peutz-Jeghersi sündroom: ennetav ravi
See on haruldane haigus, arvatakse, et selle all kannatab üks 150 000 inimesest, kuid selle diagnoosimine on väga oluline, kuna sellega kaasnevad spetsiifilised riskid ja konkreetsed protseduurid.
Peutz-Jeghersi sündroomi diagnoosimisel on suurenenud vähiriski tõttu vajalik suur onkoloogiline valvsus, seetõttu võetakse kasutusele arvukalt ennetavaid meetmeid.
$config[ads_text4] not foundIgal patsiendil peaks olema kolonoskoopia iga 2-3 aasta järel alates 18. eluaastast, lisaks gastroskoopia iga 2-3 aasta järel alates 18. eluaastast ja endoskoopiline ultraheli või kompuutertomograafia iga 1-2 aasta järel pärast 25. eluaastat.
Lisaks peaksid naised kord kuus läbi viima rindade enesekontrolli. Rindade mammograafia või magnetresonantstomograafia iga kuue kuu järel pärast 25. eluaastat, transvaginaalne ultraheli ja CA-125 määramine igal aastal pärast 25. eluaastat.
Nagu kõik, ka terved, peaksid Peutz-Jehgersi sündroomiga diagnoositud naised loomulikult oma günekoloogi juures käima Pap-määrdega.
Pärast reproduktiivse staadiumi lõppu võib naistel kaaluda emaka ja munasarjade eemaldamist.
Meestel on seevastu vaja testida munandeid ja teha ultraheliuuring iga 2 aasta tagant.
Kõik need tegevused on suunatud kasvajate varajasele avastamisele ja ravile. Mõnikord on soolepolüüpide profülaktiline eemaldamine soovitatav ka nende tüsistuste riski vähendamiseks.
Peutz-Jeghersi sündroomi, nagu ka kõiki geneetilisi defekte, ei saa ravida, kuid selle diagnoosimine on haigete inimeste spetsiifilise kliinilise ravi tõttu väga oluline.
Suurim haigusega seotud risk on neoplasmide esinemine, kuid regulaarsed kontrollid võimaldavad neid varakult avastada ja ravida. Kahjuks on kannatajatel väga suur defekti edasiandmise oht oma lastele.

















-zesp-indukowany-przez-adiuwanty.jpg)



